基于结构的的虚拟筛选(VS) 是药物研发过程中的关键步骤。其中,分子对接技术在预测蛋白质和小分子药物如何结合时起到了关键作用。传统的对接方法主要通过搜索算法探索配体的可能构象,并用打分函数来评估这些构象。这些方法为了提升虚拟筛选时的效率,对搜索算法和打分函数进行了化简用预测精度换来计算速度。然而,随着分子数据库的日益增大,传统对接方法在超大规模的虚拟筛选面前也显得捉襟见肘。为了解决这个问题,人们通过改良搜索算法、通过利用GPU的并行计算能力或深度学习算法来提升对接速度。 但目前,深度学习在保证对接速度和精度的平衡方面仍存在挑战。尽管给定口袋的分子对接在实际应用中更为普遍,但当前主流的深度学习策略主要针对盲对接,因此其应用范围受到了一定限制。大多数模型将结合构象和结合强度的预测视为两个独立的任务,使得在预测蛋白配体结合时无法同时获取结合亲和力,这对于大规模虚拟筛选不够友好。

2023年9月,浙江大学侯廷军教授、谢昌谕教授、潘培辰研究员和之江实验室陈广勇研究员团队联合在《自然-计算科学》(Nature Computational Science)发表论文“Efficient and accurate large library ligand docking with KarmaDock”,提出了一种名为KarmaDock的深度学习模型,能够快速、精确的生成结合构象并预测结合强度。并且通过后处理技术,如力场优化和RDkit构象对齐,以纠正预测配体构象中检测到的键长和角度不准确问题。 该方法的速度较传统方法提升了至少163倍,精度至少提高了22.3%。KarmaDock在针对LTK靶标的虚拟筛选中,仅耗时8.4h就完成了ChemDiv+Specs数据库(共计177万分子)的筛选,并且在筛选得到的化合物中发现了十几个经过实验验证的活性化合物。
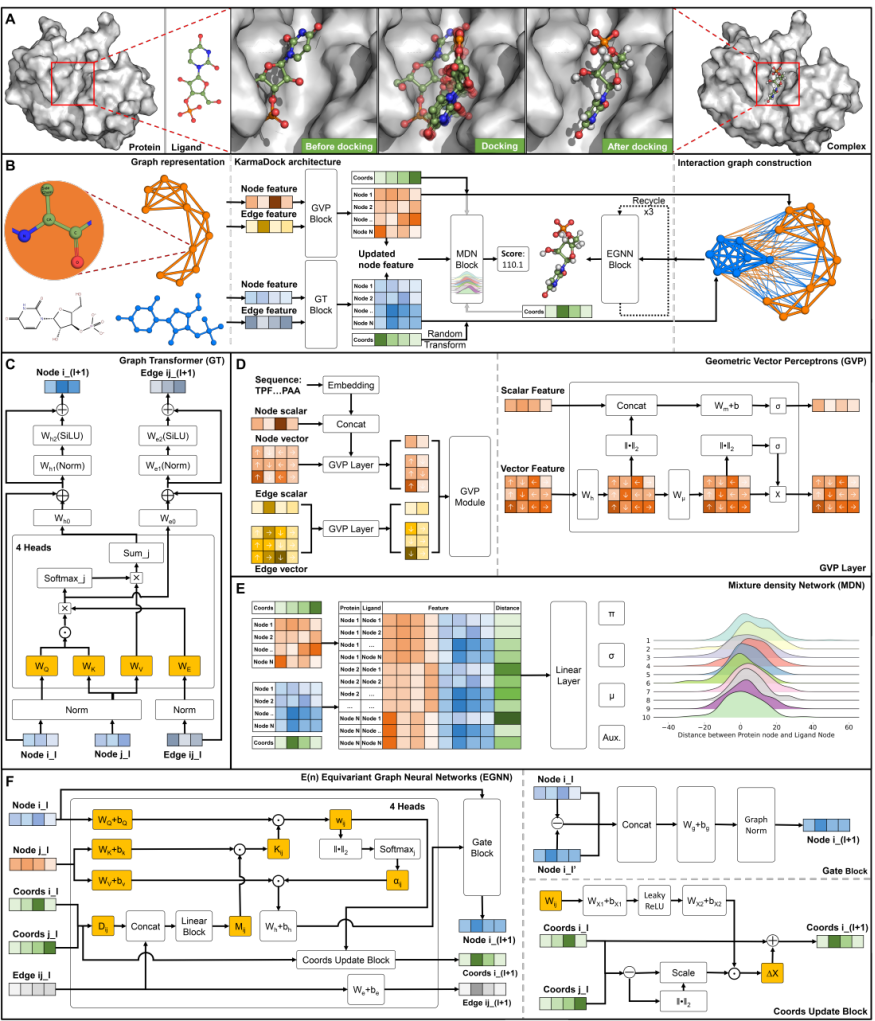
图1. KarmaDock的模型架构图
浙江大学药学院为本论文的第一署名单位,浙江大学药学院博士生张徐俊和张昊天为共同第一作者,浙江大学侯廷军教授、谢昌谕教授、潘培辰研究员和之江实验室陈广勇研究员为共同通讯作者。
原文链接:https://www.nature.com/articles/s43588-023-00511-5