PCK1 signaling pathway linked to abnormal growth of tumor cells
In the battle against tumors, researchers have made constant endeavors to unravel the enigma of the rapid proliferation of cancer cells from the metabolic distinction between normal and cancer cells, thereby curbing tumor growth through targeted therapy.
The Nobel Prize in Physiology or Medicine 1985 was awarded jointly to Michael S. Brown and Joseph L. Goldstein “for their discoveries concerning the regulation of cholesterol metabolism.” In normal cells, the “factory” of lipogenesis generates lipids “on demand”: it starts when the cell senses that the lipid concentration is insufficient and stops when the concentration returns to normal. In contrast, in tumor cells, the “factory” of lipogenesis is producing lipids on a round-the-clock basis. Even if the lipid concentration is normal in the cell, its metabolism remains remarkably active and triggers the expeditious growth of the tumor. Therefore, it has become one of the core issues to engage in research into the molecular mechanism of lipid metabolism peculiar to tumor cells.
The research team led by LV Zhiming from the Zhejiang University Institute of Translational Medicine conducted collaborative research into oncogenic signaling with researchers from Qingdao University, China Medical University in Taiwan and the University of Texas MD Anderson Cancer Center. Their findings were published online in the journal of Nature on April 8.
Metabolism basically refers to all the chemical reactions within the body used to produce energy, including carbohydrate metabolism, protein metabolism and lipid metabolism. Lipids—fats (such as cholesterol) from the diet or stored in adipose tissue (in other words the body fat)—are the “messenger” of signals.
In normal cells, lipogenesis is regulated by a negative feedback control system. When lipids reach a certain level, they will bind to INSIG proteins—anchor proteins of the endoplasmic reticulum (ER) that have two isoforms, INSIG1 and INSIG2. The binding of cholesterol-derived oxysterols is crucial for the binding of INSIG proteins to sterol regulatory element-binding proteins (SREBPs). SREBPs are inhibited by a complex composed of INSIG proteins, SREBP cleavage-activating protein (SCAP) and sterols in the endoplasmic reticulum.Regulation of the interaction between INSIG proteins and SCAP by sterol levels is essential for the dissociation of the SCAP–SREBP complex from the endoplasmic reticulum and the activation of SREBPs.
However, whether this protein interaction is regulated by a mechanism other than the abundance of sterol—and in particular, whether oncogenic signaling plays an important role—is elusive. LV Zhiming et al. focus their research on cytosolic phosphoenolpyruvatecarboxykinase 1 (PCK1), the rate-limiting enzyme in gluconeogenesis. Gluconeogenesis and glycolysis are two mutually-inhibiting reactions, with the former synthesizing glucose and the latter converting glucose into energy.
Tumor cells need to inhibit gluconeogenesis and activate glycolysis to produce ample energy. Researchers discover that tumor cells “drive away” PCK1 and force them to be “laid off”. However, PCK1 lands a new job instead of staying “at home”.
In tumor cells activated by tyrosine kinase receptors (RTKs) or the KRAS gene, AKT phosphorylates PCK1 at Ser90 and causes phosphorylated PCK1 to translocate to the endoplasmic reticulum, where it uses GTP as a phosphate donor to phosphorylate INSIG1/2. In other words, tumor cells act as a catalyst for the shift in the role of PCK1.
This phosphorylation reduces the binding of sterols to INSIG1 and INSIG2 and disrupts the interaction between INSIG proteins and SCAP, leading to the translocation of the SCAP–SREBP complex to the Golgi apparatus, the activation of SREBP proteins (SREBP1 or SREBP2) and the transcription of downstream lipogenesis-related genes, proliferation of tumor cells, and tumorigenesis. In addition, phosphorylation of PCK1 at Ser90, INSIG1 at Ser207 and INSIG2 at Ser151 is not only positively correlated with the nuclear accumulation of SREBP1 in samples from patients with HCC, but also associated with poor HCC prognosis.
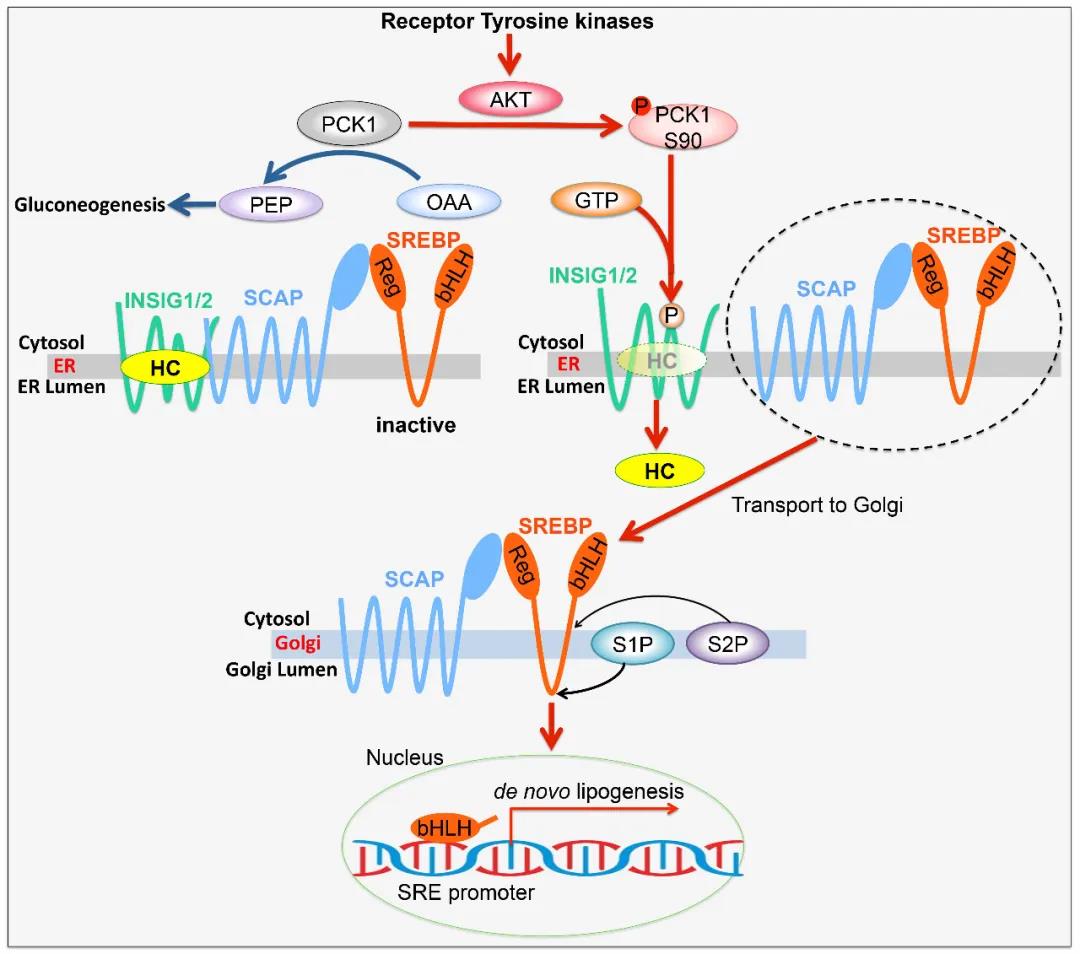
These findings highlight the importance of the protein kinase activity of PCK1 in the activation of SREBPs, lipogenesis and the development of HCC. “This study not only provides new metabolic markers and molecular targets for the customized treatment of cancer, but also offers significant guidance on the development of drugs targeting tumor lipid metabolism,” said LV Zhiming, “It empowers us to take a step further towards the understanding of tumor metabolism and find new ways to develop efficient and low-toxic drugs.”