Scientists reveal “the reproducibility crisis” in phylogenetic trees
Recently, the research team led by SHEN Xingxing and CHEN Xuexin from Zhejiang University College of Agriculture and Biotechnology and the research team led by Antonis Rokas from Vanderbilt University in the United States co-published an article in the journal of Nature Communications, revealing that ~9 to ~18% of single-gene phylogenies are topologically irreproducible.
The ability to replicate the results of a specific published experiment or analysis is a cornerstone of the scientific enterprise. In the last few years, concerns about scientists’ abilities to accurately reproduce the results of published studies in numerous disciplines, ranging from psychology and molecular biology to oncology, have steadily increased, leading to what some have dubbed as “the reproducibility crisis”. Phylogenetics, the science of reconstructing evolutionary relationships of biological entities, is fundamental to the study of biology. For example, a 2013 meta-analysis reported that phylogenetic trees in 6277/7539 (83.3%) studies published in the last few decades are irreproducible due to the unavailability of the underlying data. This study contributed to the birth of several public data repositories, such as Fighshare.
Is the information provided in public data repositories sufficient to ensure the reproducibility of phylogenetic trees? Moreover, there exist variations in terms of the phylogenetic informativeness of the underlying data (e.g., the number of parsimony-informative sites or branch support values) or the computing resources used (e.g., number of the central processing unit (CPU) cores and type(s) of processor among studies or among nodes of a supercomputing cluster). Do these variations affect the reproducibility of phylogenetic inference? What may account for the reproducibility of phylogenetic trees? What can be done to avoid the reproducibility crisis? The answers to these questions help improve the reproducibility of phylogenetic trees and provide important guidelines for software developers in phylogenetics.
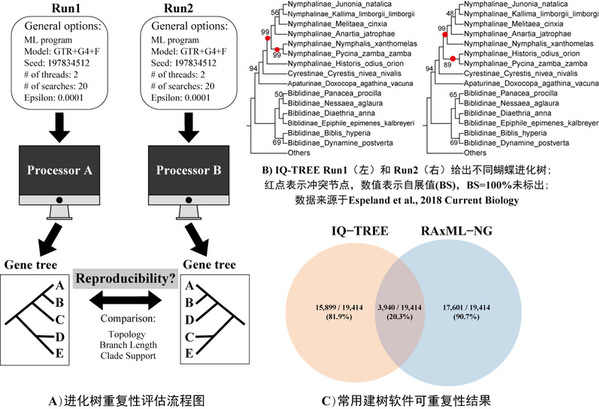
In this study, researchers executed two replicates (Run1 and Run2) for each of 19,414 gene alignments in 15 animal, plant, and fungal phylogenomic datasets. The 15 datasets comprise non-coding DNA (DNA), exon (DNA), and amino acid (AA) sequence alignments. On the basis of these gene alignments, researchers assessed the reproducibility of each maximum likelihood (ML) gene trees on the same program (IQ-TREE or RAxML-NG). For every single gene, they executed two replicates (Run1 and Run2) with exactly the same parameter settings.

Research findings indicated that 81.9% and 90.7% of the gene alignments yielded reproducible phylogneies, using IQ-TREE and RAxML-NG respectively. Only 20.3% of the gene alignments yielded topologically identical phylogenies in Run1 and Run2 of IQ-TREE and in Run1 and Run2 of RAxML-NG.
These results suggest that a considerable fraction of single-gene ML trees may be irreproducible. Increasing reproducibility in ML inference will benefit from providing analyses’ log files, which contain typically reported parameters (e.g., program, substitution model, number of tree searches) but also typically unreported ones (e.g., random starting seed number, number of threads, processor type).