Scientists report the first method to spatially deconvolve traditional bulk RNA-seq data at single-cell resolution
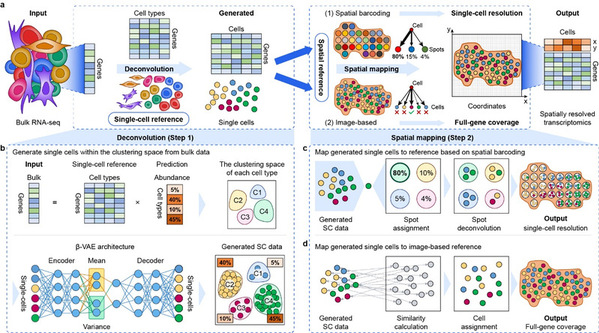
Nowadays, single-cell sequencing (scRNA-seq) and spatially resolved transcriptomics technologies have revolutionized the way people understand complex tissues and continue to drive life and health research into the era of single-cell and spatial omics. However, current single-cell sequencing and spatially resolved transcriptomics technologies are time-consuming and costly, which severely limits their application. At the same time, traditional bulk RNA-seq has long been the mainstream tool for biomedical research, with massive data resources being accumulated by many large-scale projects such as TCGA, ENCODE, ICGC, etc. Therefore, it is a major challenge for systems biology to obtain spatially resolved single-cell gene expression profiles from bulk data by making full use of the existing data resources.
Recently, the research team of Prof. FAN Xiaohui from the Zhejiang University College of Pharmaceutical Sciences, and the Innovation Center of Yangtze River Delta, Zhejiang University, together with Professor CHEN Huajun's team from the Zhejiang University School of Computer Science and Technology, and Prof. GAO Yue 's team from Academy of Military Medical Sciences, published a research article entitled “De novo analysis of bulk RNA-seq data at spatially resolved single-cell resolution” in the journal Nature Communications. The study proposed a spatial deconvolution algorithm, Bulk2Space, to reconstruct the bulk transcriptome at single-cell spatial resolution for the first time using deep learning frameworks such as β-VAE.
The research team used Bulk2Space to reveal the spatial, molecular, and functional heterogeneity of B cells in different tumor regions of melanoma and uncover spatial gene expression dynamics in different stages of inflammation-induced prostate cancer. The Bulk2Space was also applied to the spatial analysis of bulk RNA-seq data from different mouse brain regions. The bulk data were collected from Spatial-seq, a high-throughput bulk RNA-seq technology by multiplexed barcoding based on the laser capture microdissection (LCM), which was in-house developed by Professor Fan's team. The study showed that Bulk2Space can not only reconstruct the hierarchical structure of the mouse isocortex region but also reannotates ambiguous cells in the mouse hypothalamus. The benchmark test confirmed that Bulk2Space performed robustly and superior to other methods in both simulated and biological datasets. Moreover, it has been successfully applied to a wide range of biological and disease scenarios. The algorithm is now open-accessed on GitHub (https://github.com/ZJUFanLab/bulk2space).

More information: LIAO Jie (Postdoctoral Fellow), QIAN Jingyang, FANG Yin, Chen Zhuo and Zhuang Xiang (PhD candidates) are co-first authors of this study. Prof. Fan Xiaohui, Prof. Chen Huajun, and Prof. Gao Yue are Corresponding authors.
Source: College of Pharmaceutical Sciences, Zhejiang University