ZJU Scientists Discovers Mitophagy Against Ischemic Brain Injury






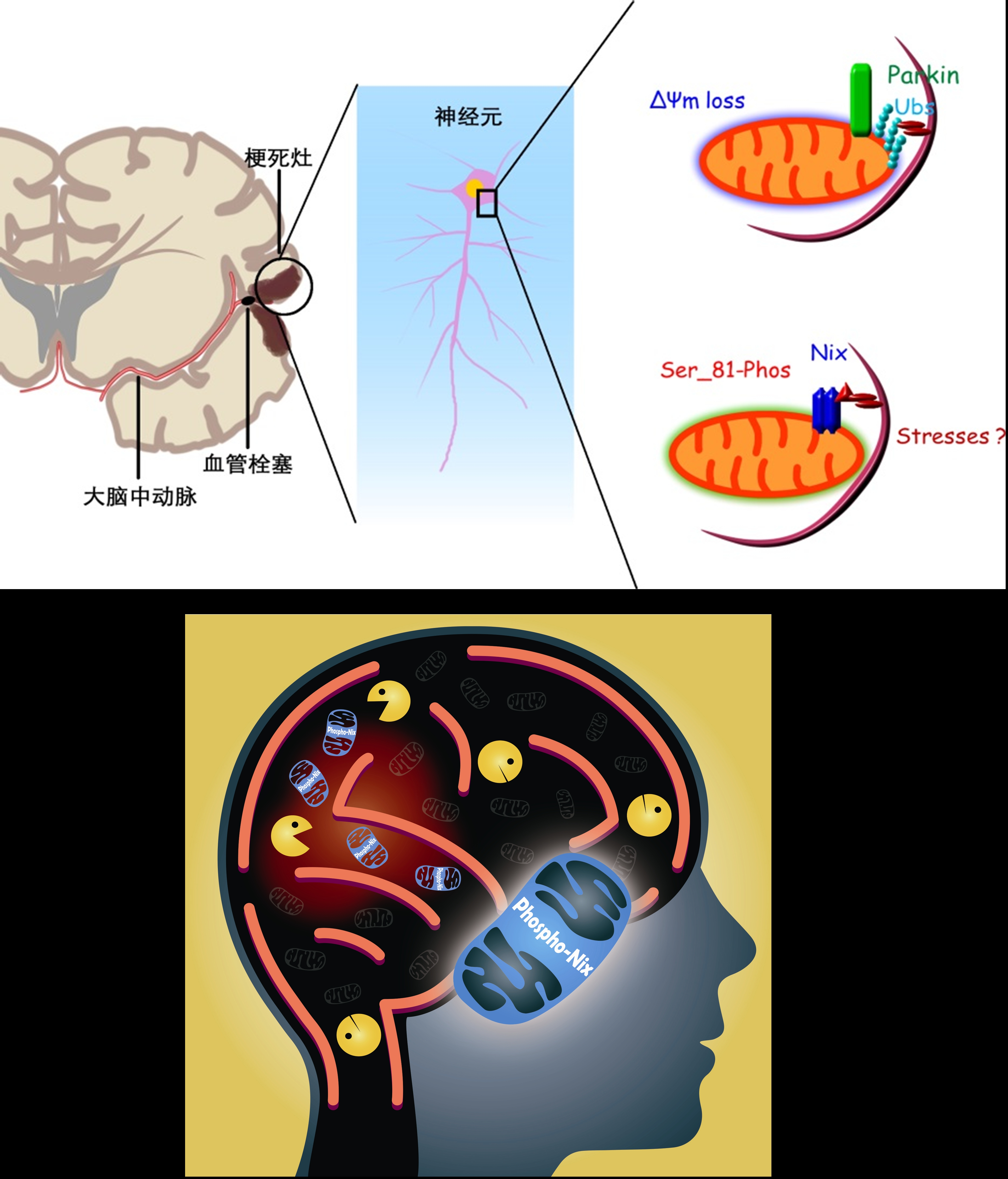
The mechanism behind ischemic brain injury is extremely complicated. Previous studies by a research team led by Prof. Chen Zhong of the College of Pharmaceutical Sciences, Zhejiang University, demonstrate that damaged mitochondria are cleared, thus attenuating brain injury, by mitophagy. However, the story behind mitophagy remains obscure, which poses a barrier to curing cerebral ischemia via mitophagy.
The current research carried out by the research team discovers the involvement of BNIP3L/NIX in cerebral ischemia-reperfusion (I-R)-induced mitophagy. Bnip3l knockout (bnip3l−/−) impaired mitophagy and aggravated cerebral I-R injury in mice, which can be rescued by BNIP3L overexpression. The rescuing effects of BNIP3L overexpression can be observed in park2−/− mice, which shows mitophagy deficiency after I-R. Interestingly, bnip3l and park2 double-knockout mice display a synergistic mitophagy deficiency with I-R treatment, which further highlights the roles of BNIP3L-mediated mitophagy as being independent from PARK2. Further experiments indicated that phosphorylation of BNIP3L serine 81 is critical for BNIP3L-mediated mitophagy. Nonphosphorylatable mutant BNIP3LS81A failed to counteract both mitophagy impairment and neuroprotective effects in bnip3l−/− mice.
The findings offer insights into mitochondrial quality control in ischemic stroke and bring forth the concept that BNIP3L could be a potential therapeutic target for ischemic stroke, beyond its accepted role in reticulocyte maturation.
Full text available online: http://www.tandfonline.com/doi/full/10.1080/15548627.2017.1357792