Scientists discover the story behind the substrate-specific recognition of IKKs






The nuclear factor κB (NF-κB), a protein complex that regulates gene transcription, is widely present in eukaryotic cells. It can specifically bind to promoters or enhancers to promote the transcription and expression of multiple genes. The NF-κB signaling pathway is closely bound up with the immune response and critical pathophysiological processes such as cell proliferation, transformation, and apoptosis. The classic NF-κB pathway is dominated by the role of IKKβ, which phosphorylates IκB family members such as IκBα and p105. Phosphorylated IκB family proteins release active NF-κB molecules into the nucleus through ubiquitin modification and proteasome-dependent degradation. IKKβ is involved in many pathophysiological processes and activates distinct substrates under different stimuli and regulatory mechanisms. However, the underlying mechanism that determines the selective substrate recognition and activation of IKKβ remains obscure.
The classic NF-κB pathway plays essential roles in innate immune responses mediated by various myeloid cells, such as macrophages and neutrophils. Activation of the NF-κB pathway is also closely related to the onset of inflammatory bowel disease (IBD). There is evidence that the intestinal macrophages of IBD patients with mutant Nfkb1 exhibit continuous activation of NF-κB signaling, and mice carrying mutant Nfkb1 that cannot express p105 present spontaneous intestinal inflammation that is similar to IBD. Therefore, the selective recognition and activation of p105 by IKKβ plays an important role in the onset of IBD. However, there is still a lack of clinical therapeutic drugs targeting NF-κB because of its widespread functions in the regulation of various physiological processes. While those drugs developed for suppressing NF-κB inhibit the corresponding pathological processes, they also have a wide range of side effects, including renal toxicity and neuropathy, and can even promote tumor progression and recurrence.
Prof. JIN Jin from the Zhejiang University Life Sciences Institute and Prof. LI Yiyuan from Southeast University co-published a research article entitled “Substrate-specific recognition of IKKs mediated by USP16 facilitates autoimmune inflammation” in the journal Science Advances.
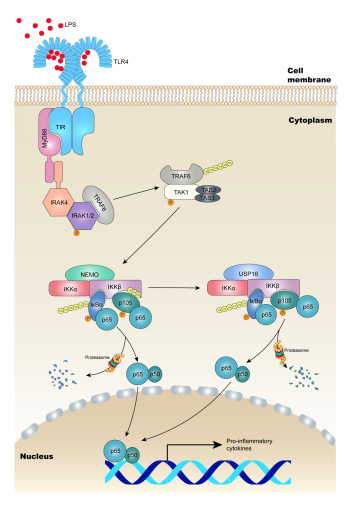
Researchers surprisedly found that p105 can still be mildly phosphorylated even without the NF-κB essential modifier (NEMO), the recruiting protein of IκBα. Biochemical data have shown that IKKβ can be modified by nonproteolytic ubiquitination during activation of the classic NF-κB signal. This ubiquitination can significantly inhibit the capacity of IKKβ to phosphorylate p105 without affecting another substrate, IκBα. By using mass spectrometry, they identified a deubiquitinase, Ubiquitin carboxyl-terminal hydrolase 16 (USP16), that specifically binds IKKα and IKKβ, and furthermore they proved that USP16 and NEMO competitively interact with IKKβ. It is known that USP16 deubiquitinates histone H2A to regulate embryonic stem cell gene expression and hematopoietic stem cell function. However, the function of USP16 in proliferation-suppressed cells remains unclear. Here in macrophages and fibroblasts lacking USP16, experiments showed significant decreases in p105 phosphorylation, while their IκBα phosphorylation remained comparable to that of their wild-type (WT) littermates. Consistently, NF-κB-targeted genes in USP16-deficient macrophages showed a clear reduction in various inflammatory cytokines when stimulated with distinct agonists of Toll-like receptors (TLRs). To further investigate the pathologic function of USP16 in vivo, a dextran sodium sulfate (DSS)-induced acute colitis model was used to mimic the clinical pathogenesis of ulcerative colitis. Mice with conditional KO of USP16 in myeloid cells (USP16MKO) displayed severe reductions in the clinical symptoms with DSS challenge. Following azoxymethane/DSS administration to establish colitis-associated colorectal cancer (CRC) model, USP16MKO mice also had fewer and smaller colon tumors than their WT littermates.
In summary, this work establishes USP16 and USP16-mediated IKKβ ubiquitination as a novel regulatory mechanism of NF-κB signaling and intestinal tumorigenesis and suggests a vital role of USP16 in colitis-related CRC pathogenesis. This study is expected to have profound implications for therapeutic approaches.